With all phenomena in the Life Sciences there are two major questions scientists ask: (1) what is the underlying mechanism (how?) and (2) what is its biological significance (why?). All eukaryotic cells replicate their DNA in a specific temporal sequence but both the mechanism of this "replication timing" program and its biological significance remain a mystery. Faithfully duplicating our DNA is arguably the most fundamental thing that our cells do, and mistakes in
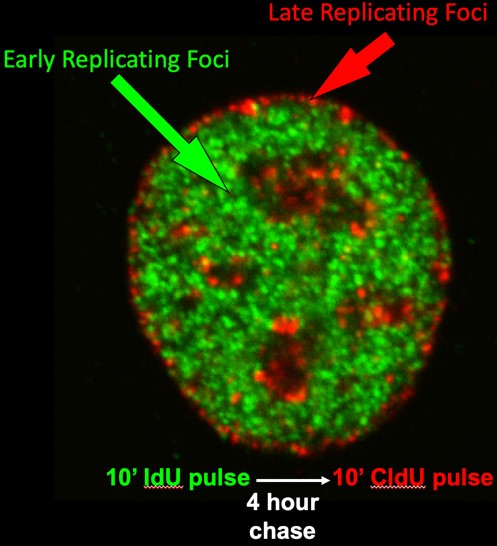
Figure 1: Cells were pulse labeled with IdU in early S phase, chased and labeled with CldU in late S phase. Immunofluorescence was performed with anti-IdU and CldU antibodies.
The primary motivation for our research is that these units that we call "replication domains" provide us with a molecular handle into the higher order structural and functional organization of chromosomes. We hypothesize that, since it is not just DNA that replicates but the entire structure of chromatin and its 3D organization, controlling where and when replication takes place can serve as a convenient means for cells to maintain stable epigenetic states and to change those states during cell fate transitions. The work in our laboratory can be divided into the following major areas:
1. Developmental Regulation of DNA Replication:
All cells contain the same genetic information (DNA) but package it with proteins into "chromatin" in characteristic ways that define each cell type. Chromatin is dismantled and re-assembled during each round of DNA replication, and we have discovered that the temporal order in which segments of DNA are replicated changes as stem cells turn into different cell types and is mis-regulated in disease. Understanding how to manipulate this packaging process may help us engineer different cell types, a central goal in stem cell therapy.

Figure 2: Replication proceeds via the synchronous firing of clusters of replication origins encompassing domains of several hundred kilobases.

Figure 3: This interactive lineage diagram has links from each cell type to data archived in the ENCODE data base. First, click the image to expand the image. Next, click on the cell type of interest, which will take you to the ENCODE data portal.
Our working hypothesis is that changes in replication timing during differentiation change the types of proteins and RNA that assemble with the DNA into chromatin and since large (half megabase) segments of chromosomes replicate as units, these changes occur across large chromosome domains. These chromatin changes would then modulate the responsiveness of genes during stem cell commitment.
Together, this body of work has provided a watershed of information that has upheld or refuted longstanding hypotheses about replication timing and generated many new hypotheses that are now testable with the systems that we have developed. We are taking both genomics approaches to evaluate the significance of replication timing to cell fate changes during differentiation, and more targeted approaches to elucidate the mechanisms relating replication timing and gene expression changes at specific gene loci.
For example, taking advantage of the information in the datasets linked to the lineage diagram in Figure 3, we were able to perform network analyses that link the expression of specific transcription factors to sets of domains throughout the genome that are regulated during liver and pancreas development and identified novel interactions between these transcription factors and their targets that may regulate the developmental changes in replication timing (https://doi.org/10.1101/186866).
2. Regulation of Replication Timing During the Cell Cycle:
Studies of DNA replication in mammalian cells suffer from a lack of systems with which to approach the problem at the molecular level. Cis-acting sequences that function as replication origins in mammalian cells have not been identified and the mechanisms that regulate where and when origins will fire during S-phase remain a mystery. Over the last 25 years we have taken a variety of approaches to this problem, revealing several discrete steps during early G1-phase that establish a spatial and temporal program for replication:
Pre-Replication Complex (pre-RC) Assembly: First is the assembly of pre-replication complexes (pre-RCs). Knowledge of this process has been greatly aided by the identification of several proteins that bind to replication origins in yeast and the identification of homologues to these proteins in higher eukaryotes. We have shown that the mammalian homologues to these proteins bind to chromatin very tightly during telophase and render newly assembled nuclei fully competent to replicate in cell-free extracts lacking these proteins.
The Replication Timing Decision Point (TDP): Shortly after the assembly of pre-RCs, a replication timing program is established that determines the order in which chromosomal domains will be replicated (Timing Decision Point; TDP).
Early cytogenetic studies by our group and others demonstrated that DNA synthesis takes place in discrete punctate foci within the cell nucleus and that foci replicating at different times during S phase are located in distinct compartments of the nucleus. We and others have shown that these foci are visual representations of stable structural units of chromosomes that replicate at specific times. We have demonstrated that the replication-timing program is established within 2 hours after the cell nucleus is formed after each mitosis, a time we called the Timing Decision Point (TDP), and that the re-positioning and anchorage of these foci within the nucleus occurs at the same time. The TDP precedes the time during G1-phase at which the sites for initiation of replication are established (Origin Decision Point; ODP), a regulatory point we discovered back in the 1990s (https://science.sciencemag.org/content/271/5253/1270.long). In fact, we can create conditions in which replication proceeds in the proper temporal order but initiates at random sites, showing that the two are regulated independently. We later demonstrated that determinants for replication timing are lost during S phase, despite maintenance of the 3D organization of chromatin, so we can say that the 3D organization is not sufficient to dictate replication timing. Thus, a central question for us is, does structure dictate function or does function dictate structure?
Taken together, our findings have led us to propose the “Replication Domain Model” (Figure 4).
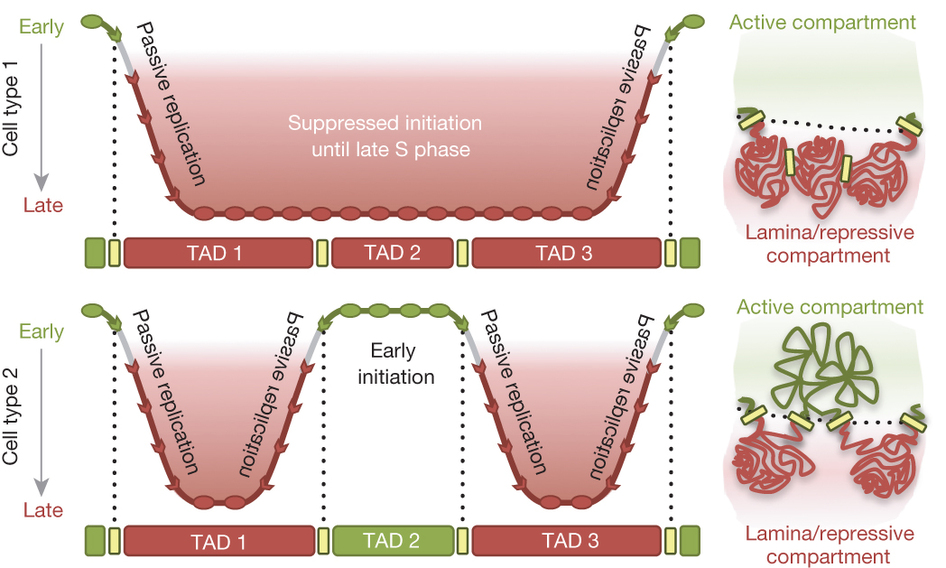
Figure 4: The replication domain model. Top left, replication timing across three TADs replicated late in cell type 1. Early initiation of flanking regions forms TTRs that extend from the left and right boundaries of TADs 1 and 3 respectively until origins throughout the late-replicating region fire. Top right, TADs 1–3 arrange in transcriptionally repressive compartments of the nucleus. Bottom left, in cell type 2, TAD2 is replicated early, creating new TTRs at pre-existing TAD boundaries. Bottom right, the switch to early replication is associated with diminished interaction with the nuclear lamina and increased interaction with other early-replicating TADs.
Our early work, mostly using microsopy, showed that replication foci (Figure 1; discussed above) are stable units of chromosome structure consisting of multiple coordinately activated replicons that retain the punctate replication pulse label for many cell generations. We showed that replication foci serve as sites of replication protein assembly and disassembly in a temporal sequence that can be uncoupled from DNA synthesis itself. We found that foci in different compartments have different chromatin composition and proposed that, since chromatin is assembled at the replication fork, organization of the genome into coordinately replicated domains could facilitate rapid domain-wide chromatin changes.
Using genomics, we discovered that replication timing consistently changes in units of 400-800 kb, defining molecular coordinates for replication domains. We showed that domain boundaries are functionally relevant in that they can confine the effects of rearrangements. We demonstrated that replication domains correspond to topologically associating domains (TADs) measured by chromatin conformation capture (Hi-C) and their higher order folding corresponds to replication timing such that domains that contact each other replicate at similar times (https://www.nature.com/articles/nature13986). Both the TADs and the higher order 3D organization is re-established shortly after the nucleus is reformed after mitosis and coincident with the TDP (above), when replication timing is established. In summary, our work shows that the way the genome is organized in 3D space has important functional consequences and provides a unifying model for genome organization that some have generously likened to the discovery of the nucleosome. We propose that anchorage of chromatin could create scaffolds that seed the assembly of sub-nuclear compartments of different molecular composition, a model that has become popular to explain many structure-function relationships in the nucleus. We have also identified a candidate protein (Rif1) that has properties consistent with a cell cycle regulated factor important for both replication timing regulation and 3D organization of chromatin.
3. Discovery of ERCEs:
The holy grail of this field since the discovery of a replication timing program in 1960 has been the identification of the control elements in chromosomes for this temporal regulation. Many investigators have tried and failed to identify them, leading scientists to propose that replication timing is not regulated by DNA sequences. With the advent of CRISPR, allowing us to make large numbers of DNA mutations in a single “replication domain”, we identified specific DNA elements that are both necessary (required in their chromosomal location) and sufficient (active in other chromosome locations) for a domain to replicate early during S phase. Consistent with our “replication domain” model, these elements are also required for the 3D structure of the chromosome domain and for localization of the domain to its sub-nuclear compartment and, we believe independently, for transcription of all genes within the domain. Consistent with our network analyses pointed out in section 1 above, ERCEs are also sites of co-occupation of transcription factors that are essential for the identity of embryonic stem cells, the cell type in which we identified these ERCEs.
This is a major breakthrough in our field. Working out the mechanisms by which these elements control structure and function of entire chromosome domains will drive research in the lab for many years to come. (https://www.cell.com/cell/pdf/S0092-8674(18)31561-7.pdf)
4. Solving the mystery of human origins:
Another one of the great holy grails in the DNA replication field is to find the origins of replication. We believe that ERCEs regulate the time at which an entire chromosome domain is ready to replicate but they are not the sites where replication begins, the “origins”. Scientists have argued whether origins are specific sites or whether replication can initiate at any site in the genome at random. Most of these scientists ignore one body of evidence or another.
We believe, based on taking ALL the evidence into account, that humans (and other organisms with large genomes) have a great deal of flexibility as to where replication can initiate and that these sites are selected stochastically (stochastic is not random, but rather with each of the potential sites of initiation being used with a different probability). As a result, each cell in a population is using a different cohort of initiation sites. Thus, we believe, all the methods that have been used to map origins that pool millions of cells to map origins are getting only an average picture, with any one site being used as an initiation site very rarely so being very difficult to detect. This has resulted in datasets that do not agree, fueling the polarization of the field.
The solution to this problem, and the test of our hypothesis, is to measure replication initiation on single DNA molecules, one at a time, in order to actually measure how frequently replication initiates at all sites across the genome. We predict that many sites, but not all, will be used to initiate repliation and that the frequency of usage will vary widely across the genome.

Figure 5: Optical Replication Mapping, DNA fibers (blue) migrating through nanochannels. Nickase sites (green) denote map positions. Red comets denote 647-dUTP incorporation and fork polarity.
We are taking two approaches to this challenge. One is to take advantage of very new technology that can stretch individual purified DNA fibers and read their map positions at an unprecedented rate – we add to that a label to tell us where replication has initiated (Figure 5). We believe that this is revolutionary technology that is poised to change how we map and study DNA replication and we intend to invest significant resources into this new approach. (https://doi.org/10.1101/214841).
In the second approach, we are trying to develop methods to map the positions of protein binding on single chromosomes. This is in the development state, but if successful, we will be able to map the sites where pre-replication proteins bind during G1 phase. This will allow us to map the specificity and degree of stochastic positioning of these “markers” that are necessary for replication to initiate and relate that to the actual sites of initiation mapped on the purified DNA fibers. This is the work of Dan Bartlett in the lab, currently unpublished.
5. Replication timing and disease:
By comparing normal and diseased cells, we can identify alterations in replication timing that are specific to disease. Two examples are as follows:
By comparing normal skin cells from individuals of widely different ages, from fetal to 96 years old we did not find significant differences in replication timing. However, premature aging diseases, which can arise from mutations in very different genes, have been puzzling because there has been no molecular commonality between these different genetic origins of the disease even though it gives rise to the same clinical phenotype. Comparing skin cells from normal individuals of different ages and patients with premature aging disease from different genetic origins revealed a set of common replication timing abnormalities: the first molecular marker of these diseases. Moreover, by converting these cells into embryonic stem cell-like cells and then re-differentiating them back to skin cells, we could re-capitulate human embryogenesis in a dish and demonstrate which of these replication timing abnormalities occur first during human development. This identified a specific gene, TP63, as a central player in premature aging disease. How the misregulation of TP63s replication timing can dramatically accelerate the process of aging is now an area of active pursuit for Dr. Rivera-Mulia in his new position at U. of Minnesota. (https://www.pnas.org/content/114/51/E10972.short)
B-lineage acute lymphocytic leukemia (B-ALL) is the most common childhood malignancy, yet we still do not understand the molecular mechanisms that lead to the genesis of this disease and there are no known strategies for its prevention. We demonstrated that pediatric B-ALL cells show alterations in replication timing from non-leukemic human B cells. Some of these alterations are linked to particular sub-types of B-ALL defined by their hallmark mutations and resemble features of specific stages of normal human B-cell differentiation suggesting that different subtypes my derive from cells arrested at these different stages. Intriguingly, some of these features were linked to whether the patients remained in remission or relapsed. Thus, RT has the potential to provide a new genre of biomarkers for diagnosis. In addition, RT reports on an undeveloped aspect of chromosome biology that is altered in B-ALL. Hence, studies of the cellular origin and mechanistic determinants of these RT alterations will provide novel insights into the underlying mechanisms of B-ALL. (https://doi.org/10.1101/549196)
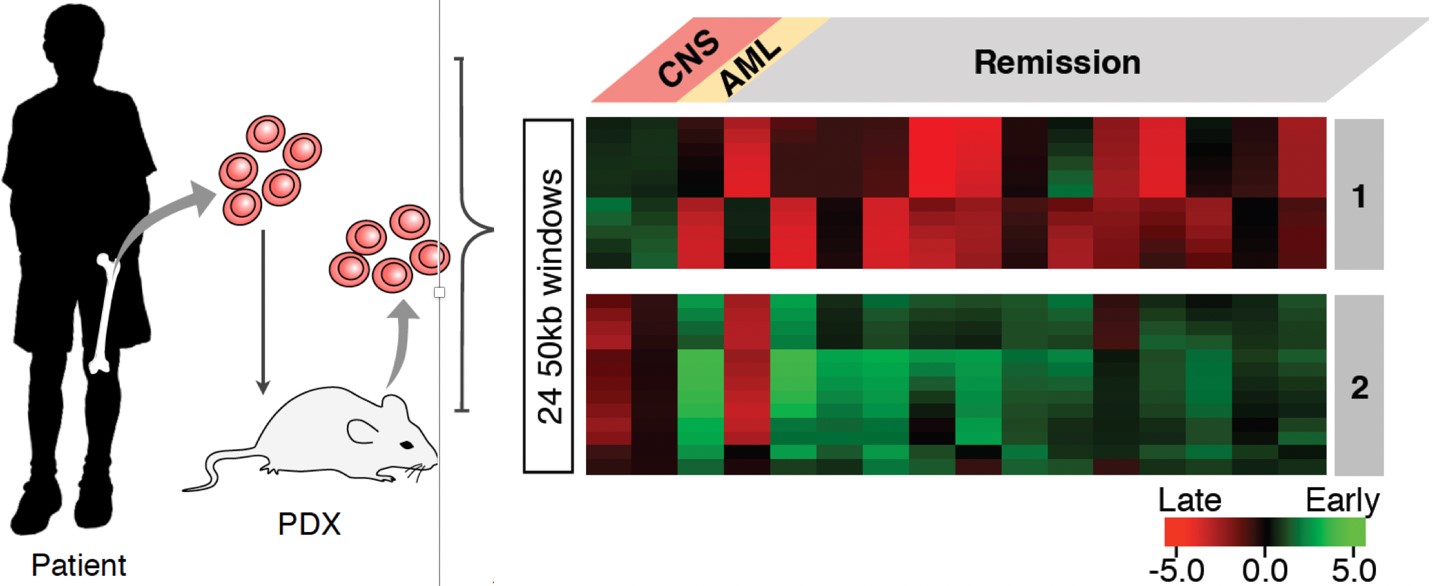
Figure 6: Bone marrow samples from children with BCP-ALL were collected at diagnosis. Children were treated with standard BCP-ALL chemotherapy. On the right are replication timing signatures from patients who either relapsed with cancer in the central nervous system (CNS) or who remained in remission several years after treatment.
